New Features
PyMOL 2.5
- Multiple-level undo for PyMOL actions
- Curved cartoon cylindrical helices
- Customizable keyboard shortcut menu
- Improved isosurface generation
- Nucleotides from the PDB can now be extended in builder
- Improved Maestro file import
- ... See https://pymol.org/d/media:new25
PyMOL 2.4
- Support for Looking Glass Holographic Displays
- Pi-Pi and Pi-Cation interactions (A > find > pi-interactions)
- WaterMap result presets (A > preset > WaterMap ...)
- APBS Plugin improvements (multi-state assemblies, propka pH calculation)
- Distinguish .mrc and .ccp4 formats (origin interpretation)
- Trajectory handling improvements
- Improved error handling in Python API with exceptions
- ... See https://pymol.org/d/media:new24
PyMOL 2.3
- Atom-level cartoon transparency
- Fast MMTF export
- Sequence viewer gaps display
- ... See https://pymol.org/d/media:new23
PyMOL 2.2
- MAE (Maestro) format export with styling, properties, and group hierarchy
- Nucleic acid mutagenesis wizard
- Stereochemistry labeling with RDKit or $SCHRODINGER backend
- ... See https://pymol.org/d/media:new22
PyMOL 2.1
-
new selection keywords "polymer.protein" and "polymer.nucleic"
-
SpaceNavigator support on all platforms
-
"auto_copy_images" support on all platforms
- ... See https://pymol.org/d/media:new21
PyMOL 2.0
- Unified modern user interface
- Anaconda Python distribution
-
Open access incentive executables
- ... See https://pymol.org/d/media:new2
PyMOL 1.8.6
-
Fast MMTF load support
-
chromadepth ray tracing support (Incentive-only)
-
smooth surface edges (Incentive-only)
-
... See https://pymol.org/d/media:new186
PyMOL 1.8.4
-
... See https://pymol.org/d/media:new184
PyMOL 1.8.2
-
cartoon_gap_cutoff: draw dashed cartoon over gaps of size < cartoon_gap_cutoff
-
auto_show_classified: When loading a file, show cartoon for polymer, sticks for organic, and spheres for inorganic molecules (Setting > Auto Show... > ... by Classification)
-
new “Distances between rings” measurement mode in Measurements Wizard
-
MOL2 file support improvements
-
... See also https://pymol.org/d/media:new182
PyMOL 1.8.0
See also https://pymol.org/d/media:new18
Open Source and Incentive
- changed defaults:
- fetch by default downloads mmCIF files (type=cif)
- scene_buttons=on
- finalize CIF/mmCIF support:
- add support for discrete=1 loading of CIF files
- improve multi-model loading (inhomogenious models, pdb_honor_model_number support)
- support loading multiple objects ("data_" blocks) from one CIF file (multiplex != 0)
- set cartoon_trace_atoms and ribbon_trace_atoms for CA-only models
- assembly setting: load biological unit
- Ignore "auth" identifiers with cif_use_auth=off (default: on)
- Display full sequence (incl. missing residues) with cif_use_auth=off (uses _entity_poly_seq)
- Bundled components dictionary subset for connect_mode=4
- added file types support: pdbqt, pdbml, cml
- API: cmd.get_object_ttt()
- Tcl/Tk window:
- Simple font size dialog (Setting > GUI Font Size)
- Feedback when changing settings
- support cartoon_trace_atoms for beta-sheets
Incentive Only
- improved mae support
- render zero-order bonds as dashed lines or sticks
- A > compute > surface area > per residue (get_sasa_relative)
- fixes stick and sphere rendering issues on OS X with ATI Radeon R9 graphics cards
PyMOL 1.7.6
Incentive Only
- Order independent transparency for real-time rendering (transparency_mode=3)
- chromadepth
- Experimental Retina resolution support in MacPyMOL (--retina command line flag)
Open Source and Incentive
-
expose "reps" and "protons" to iterate/alter
PyMOL 1.7.4
Open Source and Incentive
- faster mmCIF loading
- multi-letter chain support
- connect_mode=4
- added file types support: idx, spi
Incentive Only
- cartoon transparency, sorted in real-time
PyMOL v1.7.2 (Release Date: August 4, 2014)
Incentive Only
- Pre-integrated volume rendering: Reduces layering artefacts in volumes. It's the new default for volume objects in Incentive PyMOL.
volume_mode=0
volume_mode=1 (default) - Real-time antialiasing: Antialiasing was so far a ray-tracing-only feature. Now real-time graphics also supports antialiasing, it's disabled by default and can be activated by setting antialias_shader=1 (or 2)
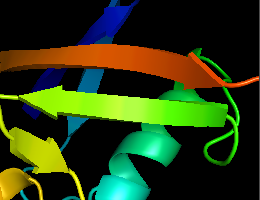
antialias_shader=0 (default)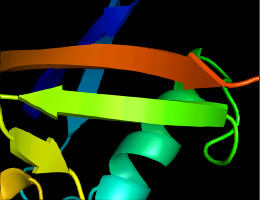
antialias_shader=2
Open Source and Incentive
- run pml and py script with load
- support ccp4 maps with mode 0 and 1
- Simple text editor to edit pymolrc (Tk GUI: File > Edit pymolrc)
- improved mmCIF file loading (load multi-model entries, read secondary structure)
- Commands
- volume now takes a ramp as its third argument
- volume_color can color volumes by custom ramps and ramp presets
- volume_ramp_new can register a new volume ramp preset
- desaturate desaturates colors
- join_states creates a multi-state object from multiple objects
- alphatoall transfers atom properties from the CA atoms to the full residue
- loadall loads all files from a file name pattern
- centerofmass calculates the center of mass
- extra_fit performs a many-to-one alignment
- api queries API details about a command
- Settings
- volume_mode to toggle between post-classified and pre-integrated volume rendering
- intensity adjustment when changing volume_layers
- antialias_shader
Bug Fixes
- fix distorted labels with stereo walleye/crosseye
- fix cartoon picking doesn't honor masking atoms/objects
- fix cyclic nucleotide was marked as polymer
- fix atom properties: assign empty string (Incentive PyMOL only)
PyMOL v1.7
Incentive Only
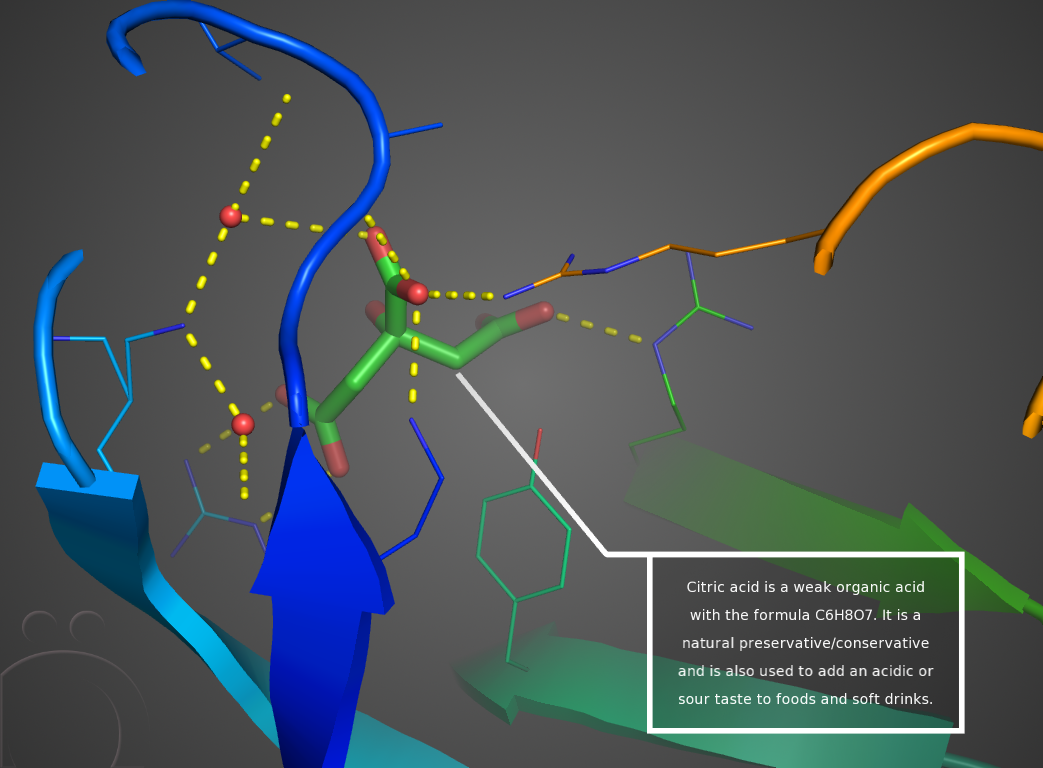
-
new additions to labels
- multi-line labels
- background colors
- connectors and outlines
-
callouts for general labeling
-
More settings added to Settings > Edit All GUI
-
Many new labels settings
-
New Search Wizard (CTRL-F) to find objects and selections in the Object Menu Panel
-
Select atoms by coordinates or user-defined properties
-
Access settings and properties from the iterate/alter commands via “s.” and “p.”
-
AxPyMOL v1.7 Released (for licensed users)
-
JyMOL v1.7 Released (for licensed users)
Open Source and Incentive
-
get_renderer command to return information about the graphics card
-
focal_blur to blur images away from the focal point
-
get_bond for retrieving bond properties
-
fetch from PubChem with SID and CID codes
-
get_version expanded to return detailed build information
-
cmd.load_coords can now take any Python array (sized Nx3) and extend an object
-
New Settings
-
pdb_conect_nodup, don't write bond-order duplicated PDB CONECT records (available in Open-Source PyMOL since SVN rev 4088)
-
session_embeds_data, to encapsulate ancillary information inside a session
-
map_auto_expand_sym, takes symmetry from map if molecular object doesn’t have symmetry information
-
- 3f atom-level settings now supported
-
Atom-state level settings
-
Color settings are now stored as colors, they can be set as either colors, hex (i.e, 0xRRGGBB), or float values ( [1., 1., 1.])
-
Suppress writing PDB “segi” field to PDB file if ignore_pdb_segi=1
-
Improved the PDB Loader GUI
-
PyMOL now reads space group from CCP4 maps
-
PyMOL treats empty space group like P1 (for best support of map formats which do not have a space group)
-
New selection keywords sidechain, backbone, metals
-
Improved and extended the Filter Wizard (Wizard > Filter).
PyMOL v1.6
Incentive Only
- Presets for coloring by representation (Color > C > by rep)
- Settings
- Over 500 settings are now documented in the Settings > Edit All GUI
- The Settings > Edit All GUI was refactored to allow window resizing, mouse wheel scrolling and searching
- Iterate and alter have improved performance
- Molecular Morphing is now easier and more complete
- Just click A > generate > morph for painless morphing
- Multi-state morphs
- "ramps" in C (color) menu
- Arbitrary object-level and atom-level properties
- Mouse wheel scrolls object-menu-panel and other scrollable widgets
- Spectrum command enhanced to take the new properties as expression
- Better tracking of hydrogens when editing with the Builder
Open Source and Incentive
- Completed OpenGL engine update; improved graphics performance.
- High-speed fonts, indicators, and 2D drawables (menus)
- Support sweep_angle setting with Program > Scene Loop Nutate/Rock
- Add bz2 support for reading PDB/MAE/MOE/... files
- Add support for width/height units in png command
- New command line argument "-y": Exit on error
- Support object=aln argument with alignto command
- VMD plugins have been updated
- New commands: split_chains, mse2met
- Spectrum command enhanced to take color lists like spectrumany
- ANISOU records are now saved if present
- Fixed alignment objects from super do not contain N atoms
- Fixed writing PDB files
- END before ENDMDL (SF-3496006)
- always right justify resn (SF-3539436)
- Fixed crash when loading with connect_mode=2
- Fixed shortcut auto-completion with exact prefixes
- Feedback fixes
- Fixed leading whitespace parsing in LITERAL parsing mode
- Fixed reading last ANISOU record from PDB file
- Surface lighting was improved for more realistic lighting
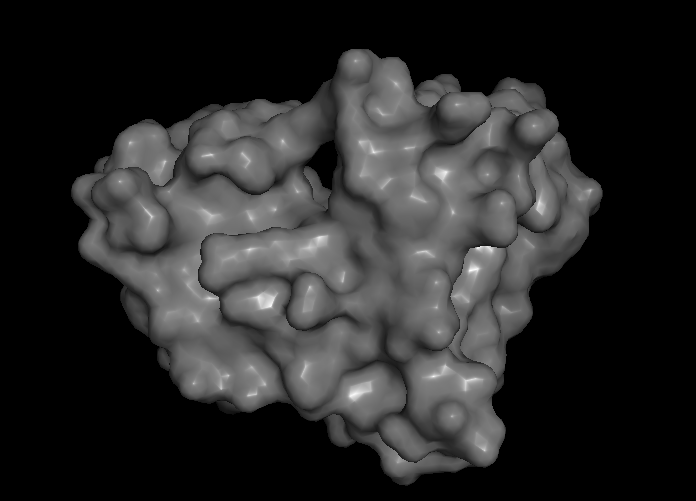
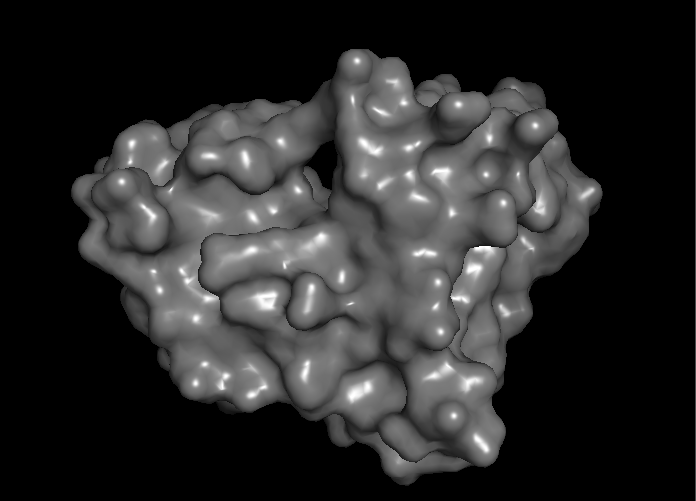
- Fixed Scripts ending on "pymol.py"
- Cealign alignment object: support for multiple alignments
PyMOL v1.5
(I) -- Incentive PyMOL only; (I+O) -- Incentive PyMOL and Open-source PyMOL
Real-time Rendering Performance
-
Increased real-time rendering performance (I+O)
-
Real-world, real-time rendering example of 250,000 atoms:
-
Lines: 28 Hz → 226 Hz (707% increase!)
-
Cartoons: 7 Hz → 47 Hz (571% increase!)
-
Sticks: 2.1 Hz → 37 Hz (1661% increase!)
-
Surfaces: 6.0 Hz → 70 Hz (1,066% increase!)
-
Real-time Rendering Improvements
  |
|
  |
|
  |
|
  |
|
-
Support for bg_gradient when ray tracing (I+O)
-
Many more non-standard nucleic acids (PSU, 5MU, 7MG, MIA, H2U, 4SU, MA6, UR3, 5MC, 40C, 2MG, TM2, 2MG) render correctly as cartoons (I+O)
-
Surface color smoothing (I+O)
-
Frame buffer-based antialiasing for real-time rendering (I+O)
-
Optimized anaglyph colors and intensities for an improved 3D viewing experience (I+O)
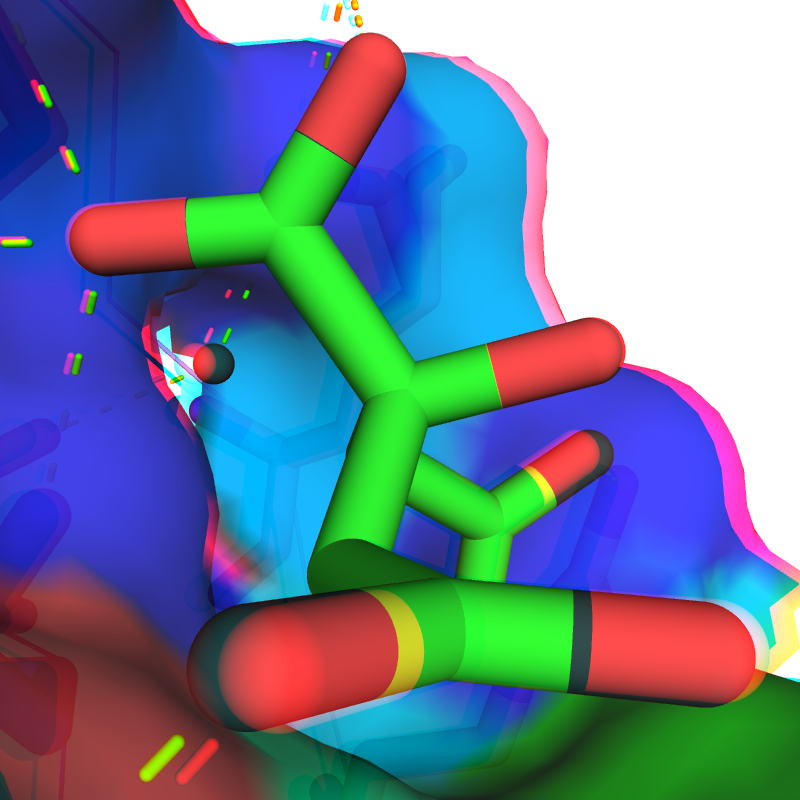 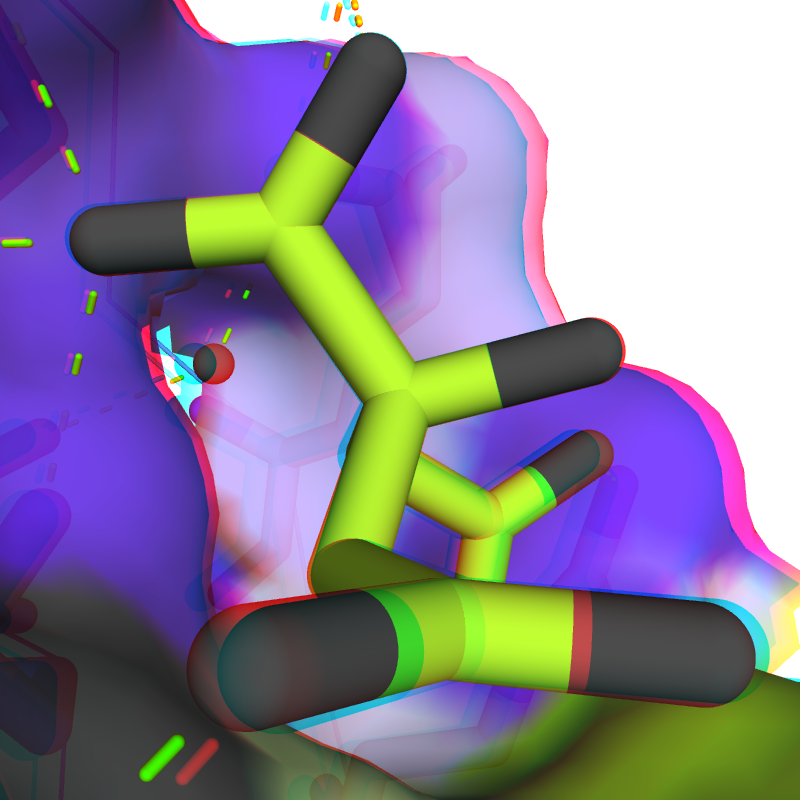 |
-
Real-time and ray-traced ambient occlusion (I+O)
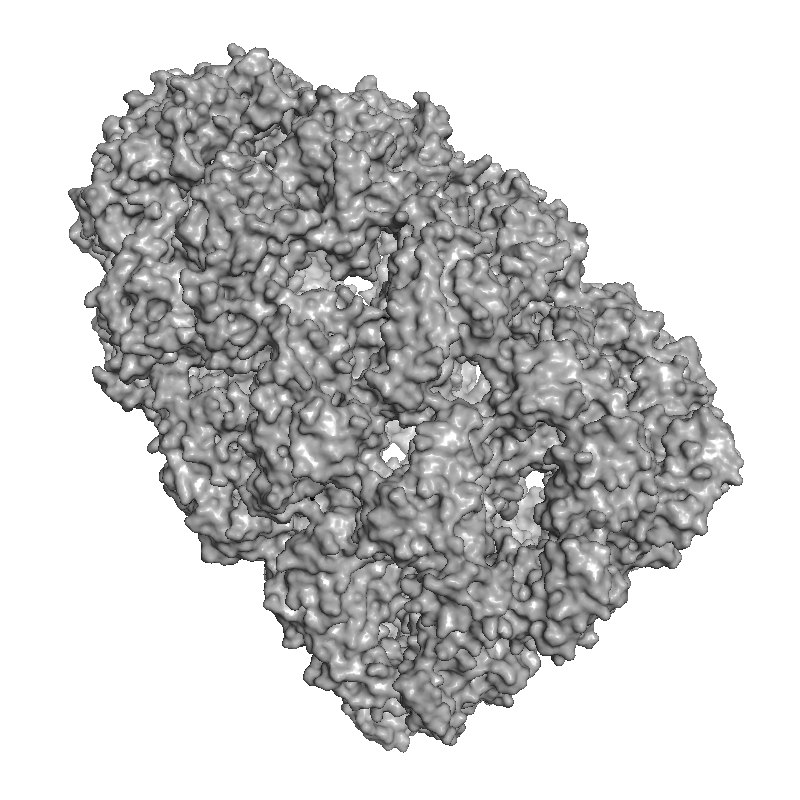 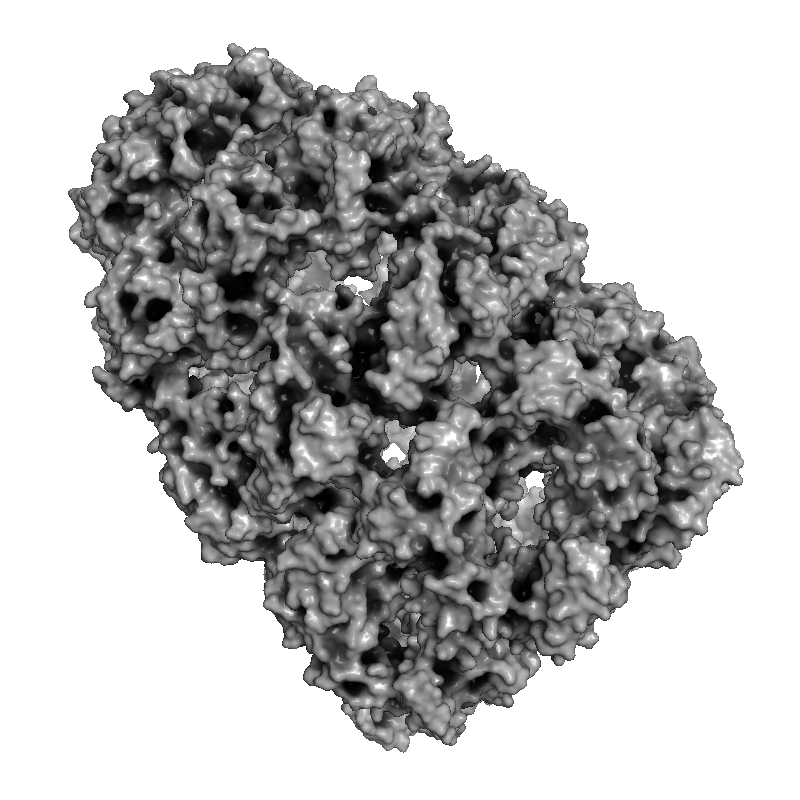 |
Molecular Modeling
-
undo now supports changes in: addition/removal of atoms and fragments, changes in atom type, valence, object creation, and more (I)
-
Improved feedback for the clean command (I)
Mac OS X
-
Support for Mac OS X 10.7 (Lion) (I)
-
MacPyMOL now comes with a Mac-friendly DMG installer (I)
-
Restored Space Navigator 3D support (I)
Other Enhancements
-
Molecular weight (A > Compute > Molecular Weight) calculation supporting explicit structure and implicit (PyMOL auto-adds hydrogens) (I+O)
-
Selection generation, A > Generate > Selection > ..., for (I+O)
- polar hydrogens
- non-polar hydrogen
- hydrogen bond donors
- hydrogen bond acceptors
- surface atoms
-
Support for reading compressed Maestro (.maegz)** and PyMOL session files (.pze, pzw, .pse.gz, .psw.gz) (I)
-
Added Show > Valence and Hide > Valence for fine-grained valence display control (I+O)
-
Added the get_viewport command (I+O)
-
Surface-based atom selecting/picking via the mouse (I+O)
-
New mouse mode for light positioning (I+O)
-
Many Fixes (I+O)
(I) -- Incentive PyMOL only
(I+O) -- Incentive PyMOL and Open-source PyMOL
PyMOL v1.4
-
New 64-bit MacPyMOL allows Mac users to access more than 4 GB of memory
-
Volume visualization for unique display of volumetric data and simultaneous visualization of multiple iso-surfaces (see https://pymol.org/volume)
-
Electron density map generation from reflection data (Mac and Linux only, Windows coming soon)
-
Stereochemical labeling
-
Improved Atom Typing; MacroModel and SYBYL/MOL2 support
-
OpenGL Shaders (GLSL) for improved on-screen rendering (cartoons, sticks, surfaces, volumes)
-
Dynamic measurements (distances, bond angles and dihedral angles)
-
File > Save Molecule now allows saving of multiple files, multi-state PDBs to a single file, and multi-state PDBs to multiple files
-
Improved State Handling makes operations on multi-state objects easier
-
Built using updated and Schrödinger-compatible distribution of Python (v2.7)
-
PyMOL Web GUI (PWG)
-
Several bug fixes
PyMOL v1.4.1
-
MTZ map reading now works on all our supported platforms and architectures.
-
Transparent ribbons – just "set ribbon_transparency, X" like other transparency settings.
-
Improved state handling makes operations on multi-state objects easier.
-
GLSL shaders now support depth cueing/fogging.
-
New background gradient. “set bg_gradient”.
-
Map resolution displayed as Angstroms, not 1/d^2, from MTZ file.
-
Elementary volume ray tracing; “set ray_volume”.
-
Cross-platform bug fixes.
PyMOL v1.3.r1
-
Fully dynamic measures
-
A > Generate > Selection > Surface Residues
-
Fixed dihedral bug
-
Added filtering step in File>Save Molecule...
-
Added mouse-mode tracking, improved neighbor finding, mixed-mode measurements to the Measurement Wizard
-
new cealign command; cealign is a new command that performs protein structure alignment using the CE algorithm.
-
updated fetch command; this command has the new capability of downloading omit maps from the EDS server.
-
new obscure command to beautifully hide proprietary data
-
many new settings for controlling measurements